cocoatree.msa.compute_normalized_seq_similarity¶
- cocoatree.msa.compute_normalized_seq_similarity(sequences, subst_matrix='BLOSUM62', gap_penalty=-4, n_jobs=1, verbose_parallel=0)[source]¶
Computes a normalized similarity matrix using a precalculated substitution matrix.
Each pairwise similarity score is normalized by the maximum possible score for the pair of sequences (i.e., the score we would obtain by comparing the sequence to itself).
Parameters:¶
- sequenceslist of str,
list of Nseq MSA sequences.
- subst_matrixstr, default=’BLOSUM62’
name of the substitution matrix. Type Bio.Align.substitution_matrices.load() to obtain a list of available substitution matrices.
- gap_penaltyint, default=-4
Penalty score for gaps. You can adjust this parameter to reflect biological assumptions (e.g., -1 for mild, -10 for harsh).
- n_jobsint, default=1 (no parallelization)
the maximum number of concurrently running jobs (-1 uses all available cores)
- verbose_parallelint, default=0
verbosity level for parallelization (see joblib doc)
Returns:¶
- similarity_matrixnp.ndarray,
a (Nseq, Nseq) array of normalized similarity scores (0.0 to 1.0).
Examples using cocoatree.msa.compute_normalized_seq_similarity¶
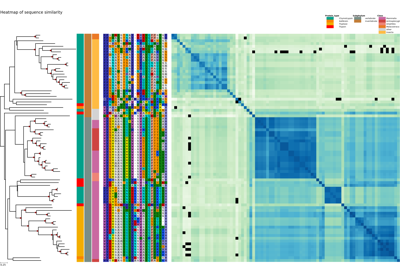
Plot a similarity heatmap of a XCoR along the phylogenetic tree