Note
Go to the end to download the full example code. or to run this example in your browser via Binder
Zooming in chromosomal interaction domains¶
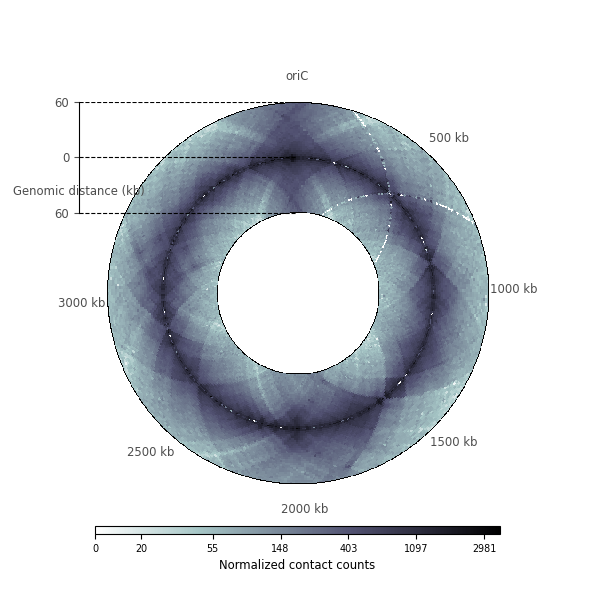
import numpy as np
import matplotlib.pyplot as plt
from circhic import datasets
from circhic import CircHiCFigure
from iced.normalization import ICE_normalization
# Load the data, compute the cumulative raw counts.
data = datasets.load_ccrescentus()
counts_raw = data["counts"]
lengths = data["nbins"]
cumul_raw_counts = counts_raw.sum(axis=0)
# Normalize the data using ICE, and keep the biases
counts, bias = ICE_normalization(counts_raw, output_bias=True)
#compute extreme values
vmax=np.max([counts[i,(i+1)%counts.shape[0]] for i in range(counts.shape[0])])
vmin=np.min(counts[counts>0])
#plotting the data
granularity = 0.5
resolution = 9958
fig = plt.figure(figsize=(6, 6))
inner_radius, outer_radius = 0.4, 0.95
inner_gdis, outer_gdis = 600000, 600000
chrom_lengths = lengths * resolution
circhicfig = CircHiCFigure(chrom_lengths, figure=fig)
m, ax = circhicfig.plot_hic(counts, granularity=granularity, resolution = resolution,
outer_radius=outer_radius, inner_radius=inner_radius,
inner_gdis=inner_gdis, outer_gdis=outer_gdis,
vmin=vmin*100, vmax=vmax, cmap="bone_r",border_thickness=0.005)
rax = circhicfig.plot_raxis()
rax.set_yticklabels(["60", "0", "60"], fontsize="small")
rax.set_ylabel("Genomic distance (kb)", fontsize="small", color="0.3", position=(0,1.03))
rax.tick_params(colors="0.3")
cab = circhicfig.set_colorbar(m, orientation="horizontal")
cab.set_label("Normalized contact counts", fontsize="small")
ticklabels = ["%d kb" % (i * 500) for i in range(7)]
tickpositions= [int(i*500000) for i in range(7)]
ticklabels[0] = "oriC"
ax = circhicfig.set_genomic_ticklabels(
tickpositions=tickpositions,
ticklabels=ticklabels,
outer_radius=0.98,fontdict={'fontsize':"small"})
ax.tick_params(colors="0.3")
Total running time of the script: (0 minutes 0.782 seconds)