cocoatree.io.load_MSA¶
- cocoatree.io.load_MSA(file_path, format='fasta', clean=True, verbose=False)[source]¶
Read in a multiple sequence alignment (MSA)
Parameters¶
file_path : path to the alignment file
- formatstring {“fasta”, “phylip”, …}, optional, default: “fasta”
format of the alignment file (e.g. ‘fasta’, ‘phylip’, etc.) All format supported by biopython’s Bio.AlignIO.read are accepted.
- cleanboolean, default=True
whether to remove ambiguous amino acids (e.g. B, X etc.)
- verboseboolean,
whether to print informations about the MSA
Returns¶
- a dictionnary containing:
sequences_id, list of sequence identifiers
alignment: list of sequences as strings
Examples using cocoatree.io.load_MSA¶
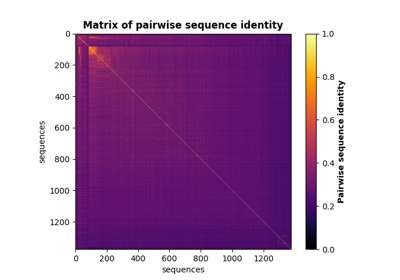
Perform full SCA analysis on the S1A serine protease dataset
Perform full SCA analysis on the S1A serine protease dataset
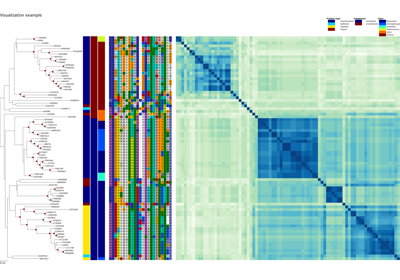
Plot XCoR together with (phylogenetic) tree and metadata
Plot XCoR together with (phylogenetic) tree and metadata

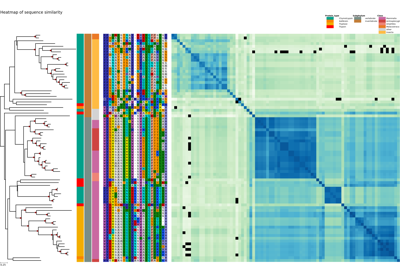
Plot a similarity heatmap of a XCoR along the phylogenetic tree
Plot a similarity heatmap of a XCoR along the phylogenetic tree